
Hello everyone, I would like to ask, how to add the following lines when creating a parameter in the rosetta for a non-standard amino acid containing a methyl group on the backbone? M ROOT 13 M POLY_N_BB 13 M POLY_CA_BB 14 M POLY_C_BB 15 M POLY_O_BB 16 M POLY_IGNORE 2 3 4 5 6 8 9 10 11 12 M POLY_UPPER 7 M POLY_LOWER 1 M POLY_CHG 1 M POLY_PROPERTIES PROTEIN POLAR CHARGED M END
When I add parameters according to the tutorial, when run script,I get the following error: python /home/tianflame/rosetta_bin_linux_2019.07.60616_bundle/demos/protocol_capture/using_ncaas_protein_peptide_interface_design/HowToMakeResidueTypeParamFiles/scripts/molfile_to_params_polymer.py --clobber --polymer --no-pdb --name X11 -k X11.kin X11.mol Traceback (most recent call last): File "/home/tianflame/rosetta_bin_linux_2019.07.60616_bundle/demos/protocol_capture/using_ncaas_protein_peptide_interface_design/HowToMakeResidueTypeParamFiles/scripts/molfile_to_params_polymer.py", line 1995, in sys.exit(main(sys.argv[1:])) File "/home/tianflame/rosetta_bin_linux_2019.07.60616_bundle/demos/protocol_capture/using_ncaas_protein_peptide_interface_design/HowToMakeResidueTypeParamFiles/scripts/molfile_to_params_polymer.py", line 1953, in main polymer_assign_pdb_like_atom_names_to_sidechain( m.atoms, m.bonds, options.peptoid ) File "/home/tianflame/rosetta_bin_linux_2019.07.60616_bundle/demos/protocol_capture/using_ncaas_protein_peptide_interface_design/HowToMakeResidueTypeParamFiles/scripts/molfile_to_params_polymer.py", line 1697, in polymer_assign_pdb_like_atom_names_to_sidechain a.pdb_greek_dist = greek_alphabet[all_all_dist[ca_index][i]] TypeError: list indices must be integers, not float
| Attachment | Size |
|---|---|
| 101.61 KB | |
| 2.79 KB |
{kind=link}
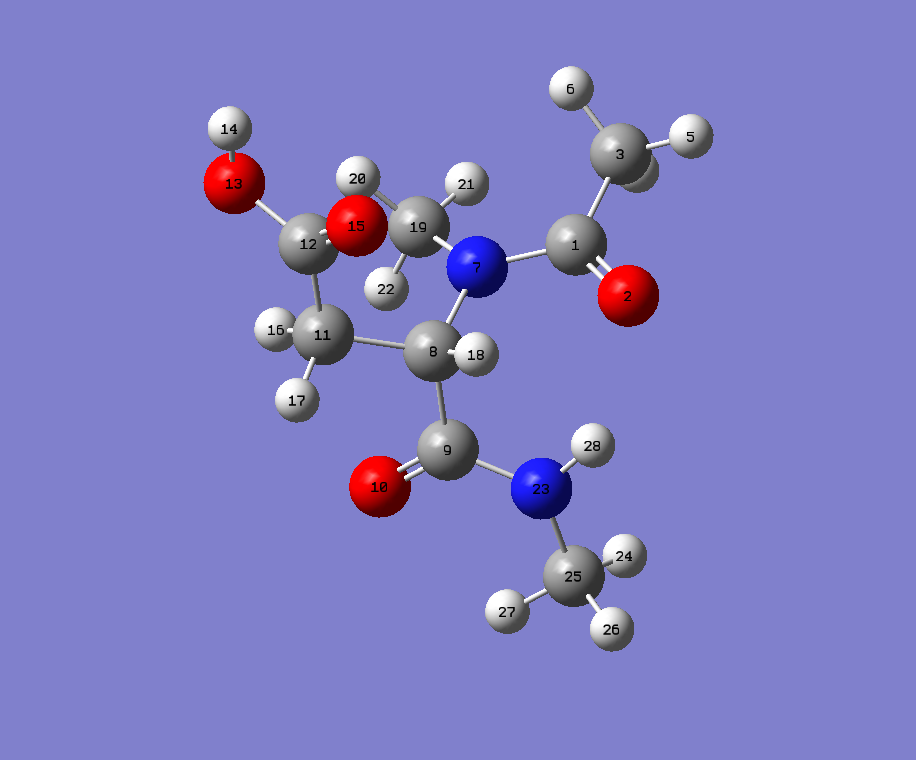
Are you using PyRosetta on Python 2, a 2to3'ed version of it with PyRosetta on Py3 or the self standing mol to params script from http://www.pyrosetta.org/scripts?
IIRC, I had some odd issues with the former (in a system with PyRosetta for Py3).
But your error seems to be that either all_all_dist, ca_index or i are floats, so simply changing the offending line to greek_alphabet[int(all_all_dist[int(ca_index)][int(i)])] might fix it. However, I have always altered the UPPER_CONNECT and LOWER_CONNECT manually or semimanually with Python+rdkit. As there are a few things that you have to change that aresn't listed like AA ASP will fail, but AA UNK won't etc.
I had to parametrise something so I checked your molecule while I was at it. And I did not get your error.
I just upload my 2to3 port of the self standing version in http://www.pyrosetta.org/scripts (old) and the version shipped with Rosetta here: https://github.com/matteoferla/mol_to_params.py just in the offchange it was a version thing...
This `polymer` flag you are using does not exist on my version(s).
There is a flag amino acid which does the same thing, but from the code I cannot see it parsing M lines. Instead it requires a Sybyl mol2 and not a sdf (MDL mol) file.
>>> babel -i mol LIG.mol -o mol2 LIG.mol2
The output will have
The names are the `
C` not the `C.2` ones... so rename manually C, CA, CB, HA, O, N.
However, do a find-replace for all `C `, `N `, `N `, `H ` names to `CX` etc. The reason being if there are more than one atom called C, the first will be called C1 during the namefixing —`CX1` etc. doesn't harm anyone, but C-prime as C1 does...
You have a methylated amine. Call the carbon there H.
Note that MDL mol files do not have partial charge, so Babel predicted it (GASTEIGER) —a bit wasteful given that you used Gaussian—, but I think this py script in turn ignores them an makes up some in turn from a dictionary.
This is what I got from your file:
Note the comedy lines:
BOND_TYPE H7 H 1
BOND_TYPE H8 H 1
BOND_TYPE H9 H 1
No idea if this file will work, but it should. But you might want to fix a few tweaks I made, such as LIG as name.
Also, is your aspartate protonated? My guess is no.
However,