
I have used ROSIE server to analyze protein-antibody interaction and predict peptide sequences of antibody that would interact with the protein.
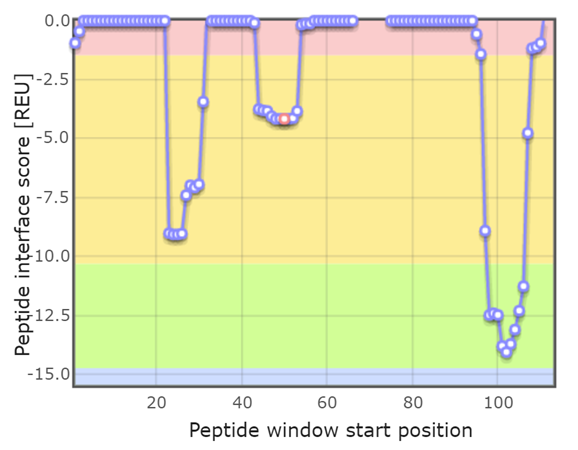
The analysis revealed 3 hotspots of the antibody sequence as in the picture attached. It did make sense to me that the predicted linaer peptides from antibody sequence were chosen from 1st and 3rd hotspots from the figure with highest contact points with the protein.
However, in cyclic peptide predictions, the server did predict only one cyclic peptide based on the middle hotspot in this figure. I do not understand the rational for that. How can the hotspot with least contact points with the protein give the best predicition for interaction upon cyclization. And why no other cyclic peptides were predicted based on the other 2 main hotspots in the interaction.
Thanks all
| Attachment | Size |
|---|---|
| 68.21 KB |
{kind=link}
The most likely reason is that there is no good way to cyclize the other two linear peptides. Not all linear peptides in bound complexes are compatible with cyclic geometry.
That being said, there are many ways of cyclizing peptides, and to my knowledge, Peptiderive uses a very narrow definition of "compatible with cyclic geometry": it only considers cyclization schemes involving disulfides between terminal residues of linear peptides when the terminal residues are already close enough to gether to make disulfide closure likely. (See Sedan et al. (2016). Nucl. Acid Res. 44(W1):W536-41. doi: 10.1093/nar/gkw385. ) If you want to try more exotic approaches, including N-to-C amide bonds and linkages that pass through many amino acids (possibly including D-amino acids), start with the linear peptide produced by the ROSIE server and try cyclizing using the methods described in:
Bhardwaj, Mulligan, Bahl et al. (2016) Nature 538(7625):329-335. doi: 10.1038/nature19791.
Dang, Wu, Mulligan et al. (2017) Proc Natl Acad Sci U S A. 114(41):10852-10857. doi: 10.1073/pnas.1710695114.
Hosseinzadeh, Bhardwaj, Mulligan et al. (2017) Science 358(6369):1461-1466. doi: 10.1126/science.aap7577.
You can use the simple_cycpep_predict application (https://www.rosettacommons.org/docs/latest/structure_prediction/simple_cycpep_predict) to determine whether a given design uniquely favours the binding-competent conformation, or whether there'd be a high entropic cost to ordering a disordered peptide on binding.
Thanks. It makes more sense now. Your feedback was very helpful.