
Hi,
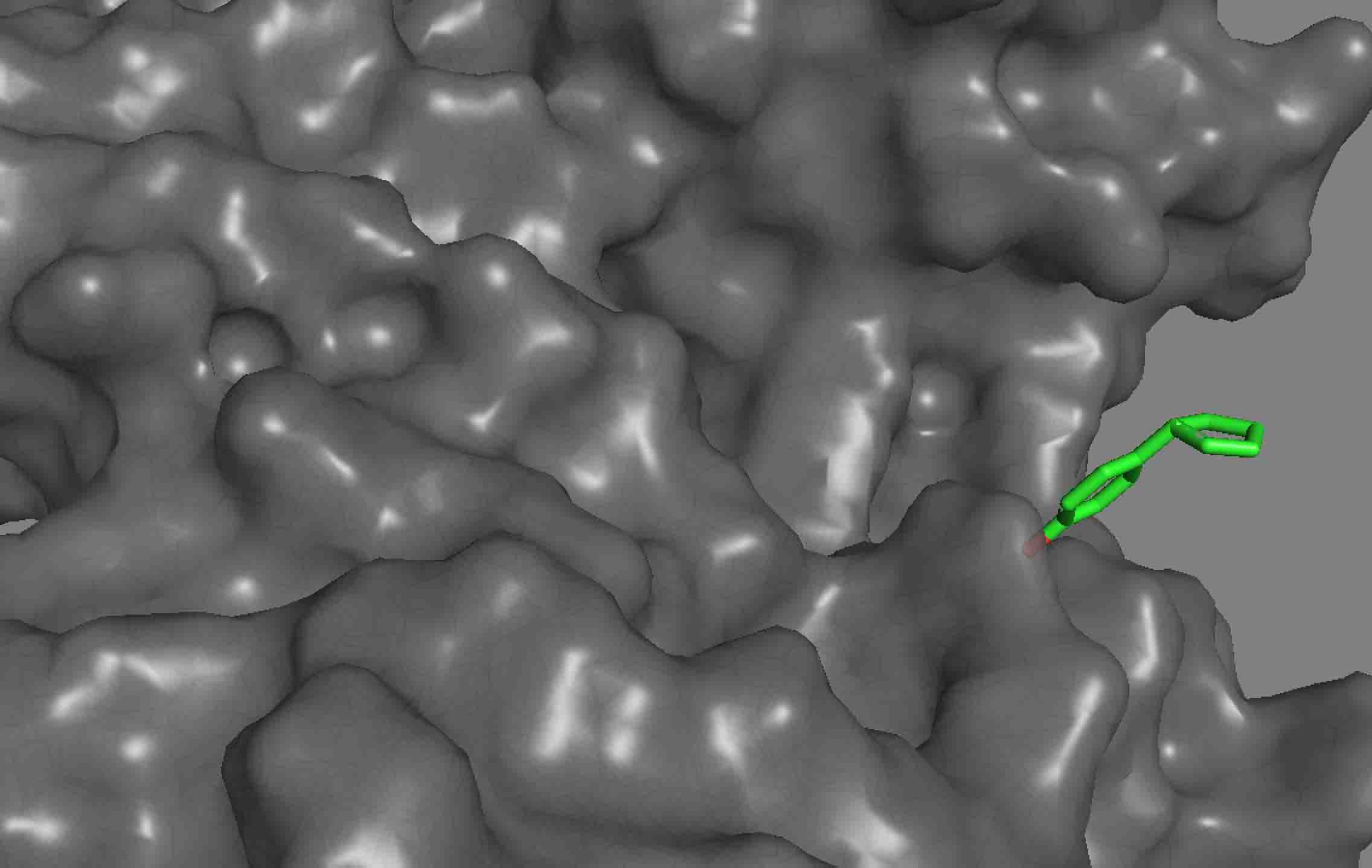
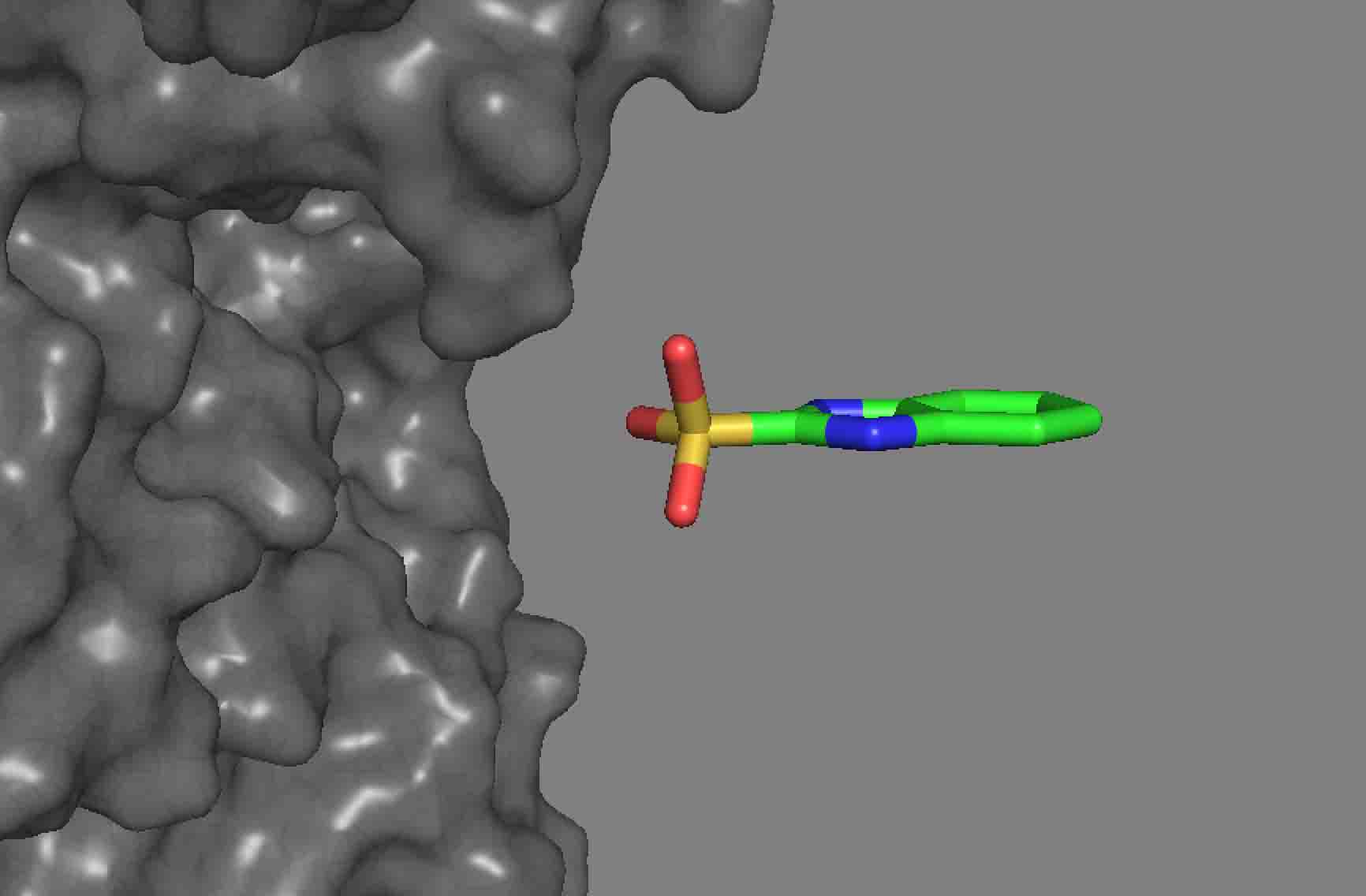
I have been trying to do ligand docking using the tutorial provided here (https://www.rosettacommons.org/demos/latest/tutorials/ligand_docking/ligand_docking_tutorial) and try to dock some of my molecules onto a protein. Before docking, I relaxed the receptors. The dock does not place the ligands into any pocket. It just keeps it floating on the surface. Also, for some of the molecules, the docking does not even happen. Please see the pictures attached. I used the exact same parameters provided in the tutorial. Is there a way to make it look for pockets globally and tune anything so that the protocol can find deep pockets? We ran the same simulations using Autodock and the autodock actually docked the ligands into the pockets.
| Attachment | Size |
|---|---|
| 71.17 KB | |
| 48.6 KB |
{kind=link}
{kind=link}
Category:
Post Situation:
The RosettaLigand docking protocol doesn't have any pocket detection logic -- it relies on you to start the ligand in approximately the correct location. (Normally within 5 Ang or so.) If you start your ligand in a random location (which is effectively what you're doing if you try to dock without specifiying a starting location), Rosetta will attempt to dock at that location, which may be quite far from the pocket you want, or even quite far from the protein.
What you'll need to do is either take the ligand in your starting structure and manually move it to approximately the location where you want it to be docked, or use the StartFrom mover from within the RosettaLigand XML to specify where it should be docked.
With the most recent weekly releases, there's facilities to do a multi-start ligand docking, where you can specify multiple locations. This way you can set up starting locations across the surface of the protein and let Rosetta search all of them. (Note you'll get results from each starting location -- you'll need to filter your output results to pick only the top structures. But that's something that you'll be doing anyway - you just need to adjust the stringency.)