
Hello,
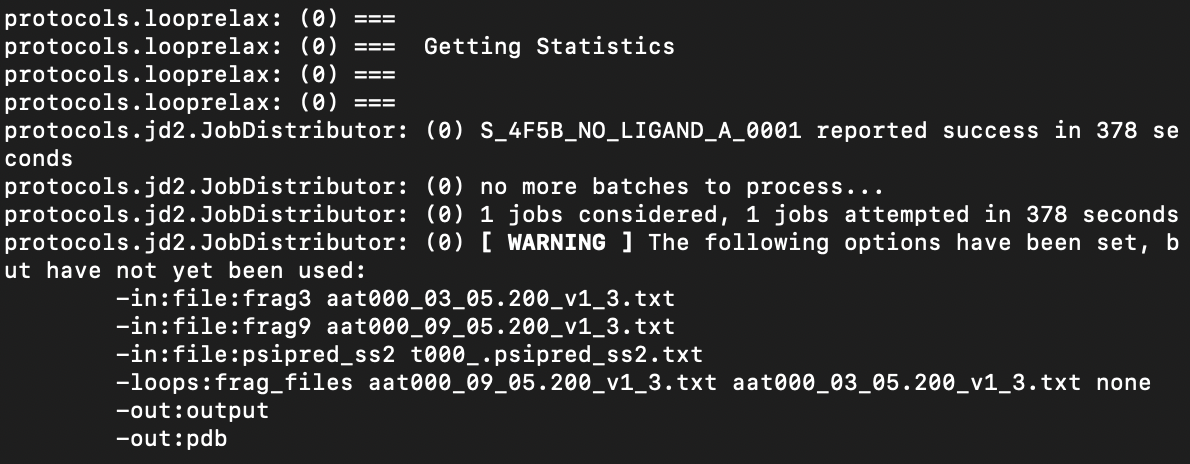
I am new to Rosetta 3 and trying to do some homology modeling of my mutated protein to a known template PDB. I have been using the minirosetta script with accompanying flags file. It appears to function correctly, though some errors pop up regarding debuging the fragment files generated by Robetta. Unfortunately, the output files only inlcude the threaded_model.out and threaded_model.fasc. There is no PDB file available for viewing in PyMol to determine the quality of the structure prediciton. I have attached a screen shot of the final information following the running of minirosetta and the flags file used. Please any help regarding this matter would be appreciated.
Justin
| Attachment | Size |
|---|---|
| 2.15 KB | |
| 304.81 KB |
{kind=link}
Category:
Post Situation:
Due to the number of structures normally generated, it defaults to outputting a Rosetta silent file. And due to the age of the protocol, it doesn't use the same input/output machinery that most of the other applications in Rosetta does, so adding the -out:pdb flag doesn't change that.
What you should do is use the SCORE line information to figure out which of structures in the silent file you're interested in (I guess it would only be the one, given that it looks like you're doing an -nstruct 1 test), and then use the extract_pdbs application (with -in:file:tags or -in:file:tagfile) to extract just those models you're interested in to PDB. (The vast majority of the structures you get from this protocol will not be of interest and can be ignored.)
By the way, the -run:constant_seed and -run:jran options are mainly used for testing and debugging. For actual production run purposes, you probably want to remove them. (Or if not, be dilligent about changing the jran numbers between different runs.)
Also, it looks like you're using the old loop-remodeling based homology modeling protocol. That protocol really isn't used at all anymore by people in RosettaCommons community. Everyone has moved over to the RosettaCM protocol (even for single-template cases). See the Rosetta documentation for details, and http://meilerlab.org/index.php/rosetta-tutorials for tutorials on how to run RosettaCM.
Thank you for the quick resposne and help. I will look into the comparitive modeling protocols.