
Dear community,
I am new to MPI so please correct any misuse of terms. My question is about optimizing for MPI on an HPC or a second (preferred) solution to my issue is to optimize my RAM usage so I don't have to run MPI, serial would actually improve overall models produced anyways.
First I have a very simple question, in the output of rosetta how do I know how many cores are being used in the MPI? I see (0), (1), and (2) at the beginning of each line so I'm assuming this mean 1 2 and 3 cores being used for that output line, is this correct? Basically if I'm using 2 cores with MPI in the output how do I know they actually got used?
I'm trying to optimize my rosetta MPI run on an HPC and my code currently is running 3X slower than expected and from reading documentation I think it just might be an option incompatibility so hoping someone with experience can advise. I have a lot of experience running with HPCs charging by the CPU but I'm using stampede2 which charges by the node so things get complicated here. Normally I'd prefer to run completely in serial but the maximum RAM the node has is 96GB and my system uses 3GB per run limiting me to about 35 runs per node (emperically tested this) but I have 68 available cores thus the want to include MPI to take advantage of the 36 idle cores (assuming I play it safe and only run 32 runs/node). Again with the HPC I'm using you can only request an entire node not individual CPUS.
So this is my plan of attack:
Request 1 node which has 96GB of RAM and 68 Xeon PHI CPUs. run 32 independent jobs to prevent exceeding RAM but each job will have 2 CPUs working in MPI.
programs I'm using to submit (screenshots attached):
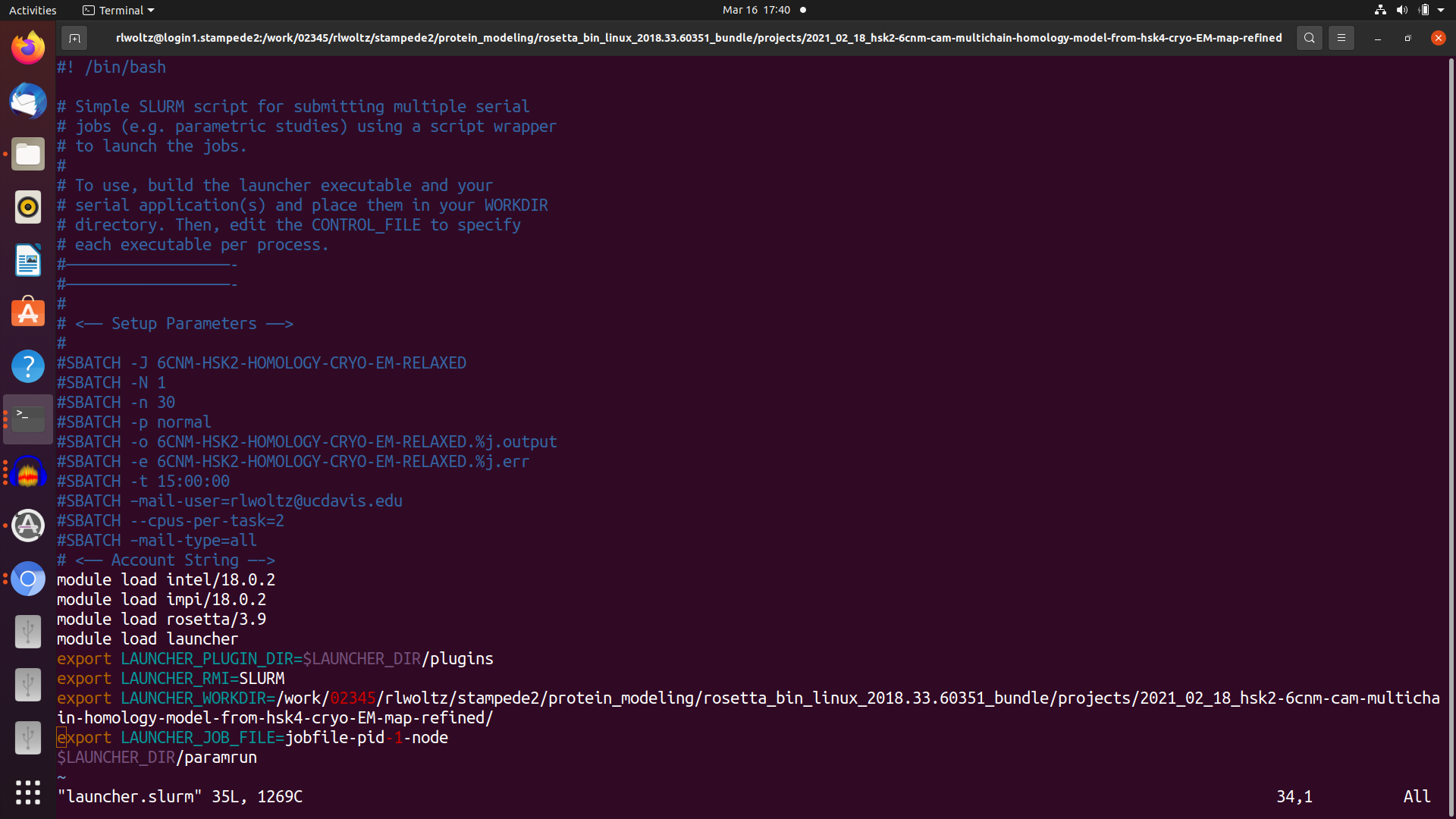
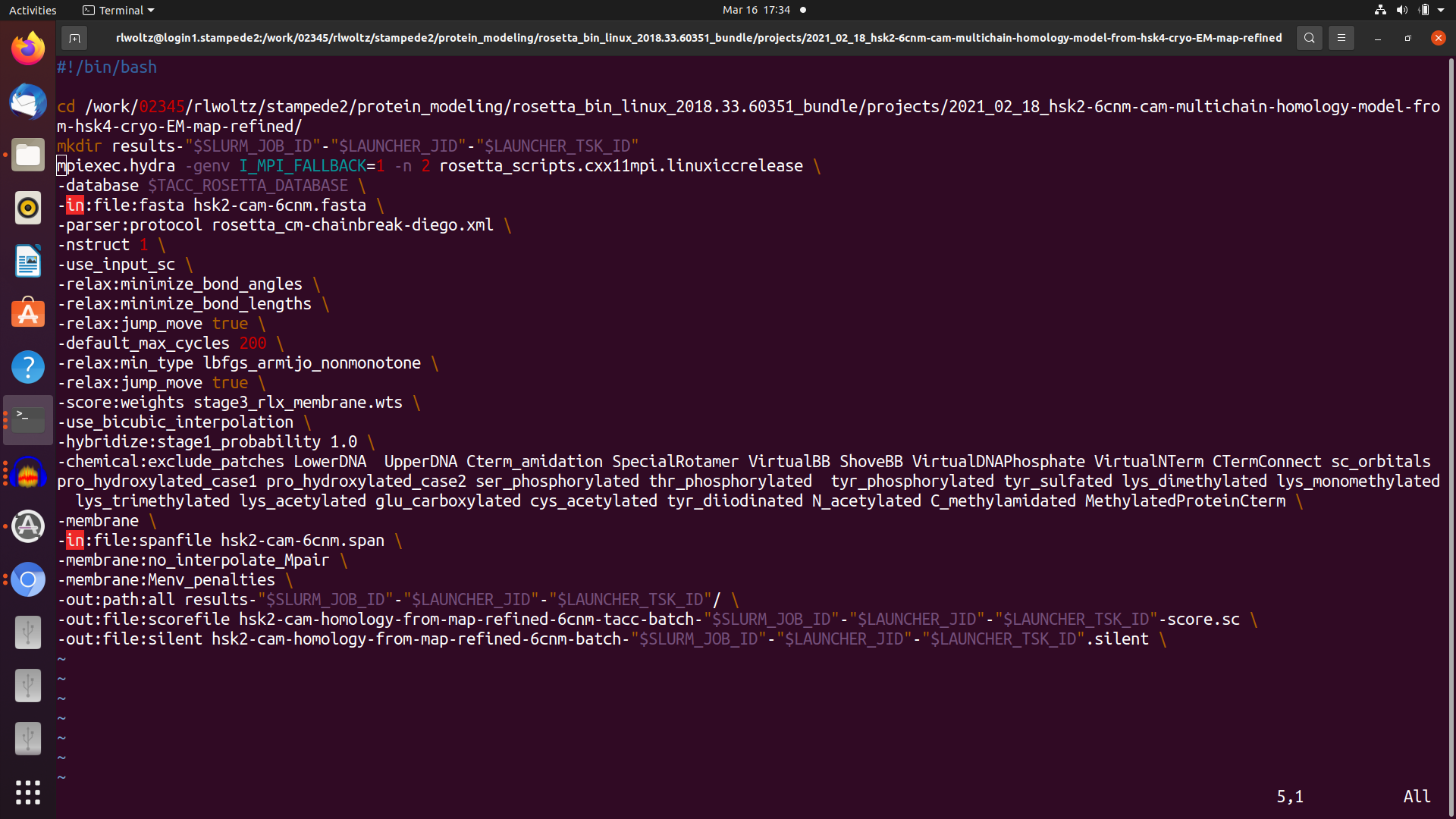
I have 4 scripts that pass things along. First I have my sbatch submission script which uses the launcher module. The main objective in this is to request 32 tasks to be run at a time with 2 CPUs per task. Then I have a jobfile that essentially lists the total number of commands (tasks) I'd like to run, for example 320 tasks would run 10 rosetta runs on 32 (X 2 for MPI) CPUs in parallel and independently. This job file runs the rosetta command file which must use mpiexec (not mpirun) command explained below. Finally this also calls the rosetta.xml but this doesn't really play a role in the sbatch but it is there for completeness.
Double checking run:
When checking top on the node the job is submitted to 65 CPUs are in use (64 for rosetta jobs and 1 for node managment?). However, I do notice that there are 2474 total, 61 running and 2413 sleeping so not sure what this refers to maybe threads (NOTE: these numbers are for my latest 30 CPU test not 32 as I'm describing)? Unfortunately, top is the only montoring program (I use htop) so if there is a command that can be used to officially check the CPUs being used I'd appreciate it. also in the rosetta output it has a (0) and a (1) for each line. I'm assuming this refers to 1 or 2 cpus being used but please clarify if I'm wrong. When I tested with 4 CPUs the max number I saw was (2) so maybe a solution is to run 22 tasks and MPI CPU=3?
output files (don't think it is helpful for troubleshooting but might be helpful in advising where to ask me to start looking):
Jobfile also has the rosetta terminal output saved with a file embedded with a unique stampede2 job id, launcher job # (1-320), and a launcher task id which I believe is the CPU id number running the script. Each Rosetta output silent files also has these unique embedded codes and saves all silent files to unique results directory.
The problem:
So everything runs and I believe it is doing MPI in the way that I'd like it to but don't know how to check other than the (x) line mentioned above. But it is very ineffecient and takes about 13 hours versus 4-6 hours per model on my laptop. I'm doing a homology model with symmetry, membrane, and loops of a protein that is 2100 AA in total and I've spent a lot of time optimizing this part. My preference obviously is to run completely in serial but because of RAM limitations this isn't possible. I'd assume the MPI would be faster, not 3X slower, than serial so I'm just wondering if I can optimize the options here. I read this about "bind to none" option in openmpi but again I've never dealt with MPI and so the terminaology is difficult for me to understand. Again in the top there are a lot of sleeping tasks so this is why I think it could be optimized. I've attached screenshots of the slurm submission script, the jobfile script, and the mpiexec rosetta run script.
Possible solutions:
I've played around with options but because of the queue and the 13 hours to make a model it is taking days-weeks to get any meaningful results. I did test with a shorter 5 min run but it didn't scale. I'm continueing to try new things but thought I could save time by asking for advice. I have a feeling it has something to do with either the launcer slurm script or the way I'm calling for MPI. The launcher slurm script calls for -n = 32 # number of tasks ---cpus-per-task=2. In the rosetta MPI running file I have this line:
mpiexec.hydra -genv I_MPI_FALLBACK=1 -n 2 rosetta_scripts.cxx11mpi.linuxiccrelease \
-genv I_MPI_FALLBACK=1 #must be included otherwise the job fails. Something with Fabric and fallback disabled so don't understand this bit but included because it works.
-n 2 #number of cpus per task? but the document I read suggested this is how many time the command is run so I'm confused by this, maybe it should be 32?
There is also this passage:
Please note that mpirun automatically binds processes as of the start of the v1.8 series. Three binding patterns are used in the absence of any further directives:
- Bind to core:
- when the number of processes is <= 2
- Bind to socket:
- when the number of processes is > 2
- Bind to none:
- when oversubscribed
- It sounds like I should bind to core since I'm running with 2 but really I'm running with 32X2 so should I bind to none?
- MPIrun document:
- https://www.open-mpi.org/doc/v3.0/man1/mpirun.1.php
- Sorry if this isn't a problem so much as helping me improve my code and unfortunately no one in my network has experience running rosetta in MPI.
Thank you for your help and please let me know if more information or clarification is needed,
Ryan
| Attachment | Size |
|---|---|
| 287.08 KB | |
| 491.5 KB | |
| 310.21 KB |
{kind=link}
{kind=link}
{kind=link}
I have not read this entire post but I am going to offer a few comments that may help.
0) Rosetta does not use MPI for intra-job parallelism. MPI is used only for embarassingly-paralell parallelism (one of those is spelled wrong). Throwing more processors at most Rosetta jobs will give you more outputs per block of time, but the absolute wall clock time of those units will not improve: it will make two structures independently on two processors in X time units, not one structure on two processors in X/2 time units. It also doesn't improve RAM usage, every Rosetta process uses the same full RAM footprint. If your cluster jobs are slower than your laptop jobs, either you are hitting virtual RAM heavily or your cluster processors are slow. (could be either...GCE is slower than my physical servers because it's just slower.)
Go-faster-per-structure, use less memory is available in the multithreading build not the MPI build. That does not scale perfectly to processors the way MPI does, it does scale better to RAM the way you appear to want, and it works for only a handful of things. I don't know what those things are or how to use it. I think it works for packing, though; unsure of minimization. (If it works for those it works for HM).
1) how many processes does Rosetta use?
This depends on your job distributor choice and your flag use. rosetta_scripts uses JD2, which is the common use case. By default, JD2 uses the "work pool job distrubutor" which devotes the processor with rank 0 to arranging work and processors with positive rank to doing work. a job with 3 processors is spectacularly ineffeicient, as 1/3 of the processors does almost nothing. use larger jobs or use the "work partition job distributor" flag instead (I can't remember what it's called, but -work_partition_job_distributor is close) will help if you want to stay at 3. That flavor of job distributor does no MPI communication at all; it just uses the mpi rank and count to divide up the jobs list evenly among processors.
Thank you for your quick reply and sorry for the delay, wanted to test out the multithreading approach. Yes I did make that post long so apologies but I wanted to include as many details for trouble shooting as possible. Correct me if I'm wrong but it seems the MPI does what a multiple initiated job will do but just in a cleaner way. I'm mostly posting this for resolution for anyone in the future looking for an answer.
I did test my job on multithreading and unfortunately it appears that the it is failing possilbly at the minimization step. Maybe you'll know what step this is referring to but my script does include a lot of parts of HM. Symmetry, membrane, loop, and multiple chains. Here is the error:
ERROR: The Membrane_FAPotential is fundamentally non-threadsafe, and cannot be used in a multithreaded context.
it did say I could contact Rebecca Faye Alford but the script did not use multiple CPUs up until this point anyways and the computer is about 10X slower than my laptop and 3X slower than the local cluster so I think I'm going to see if I can transfer my allocation elsewhere. Stampede2 does not seem like an optimal computer for large protein modeling on rosetta due to the limited RAM per node. It would be a good resource is the jobs are small in RAM usage or if their modeling is multithread safe and they can get multiple CPUs to work on the same problem. I followed this document to get my multithreading installed and running.
https://new.rosettacommons.org/docs/latest/rosetta_basics/running-rosetta-with-options#running-rosetta-with-multiple-threads
Thank you for your help in confirming this,
Ryan