
Hello,
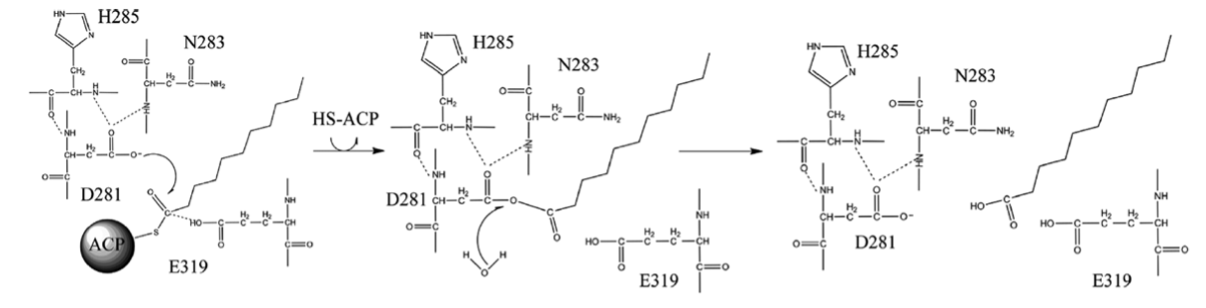
I am modeling an enzyme and a substrate to study the enzyme specificity. From literature, I learned that the specificity is determined by the second step of the mechanism in which the ligand is covalently bound to D281 of an enzyme (see the attached picture showing the mechanism). Is Rosetta able to do covalent ligand docking? If so, how can I parameterize the ligand (which is covalently bound to the protein)? If not, can I do covalent docking with other software(??) and then score it with Rosetta?
Any suggestions/papers in this regard is really appreciated.
| Attachment | Size |
|---|---|
| 87.92 KB |
{kind=link}
Category:
Post Situation:
Yes. I did it en mass and it is doable. However, I do not recall how one makes a covalent with the official topology generation script (molfile2params.py), so cannot give advice there.
I got fustrated by molfile2params.py, so I wrote my own — rdkit_to_params module for Python3 (pip released, Rdkit-to-params repo). I also made a web interface, https://params.mutanalyst.com/ which is rather rudimentary (one weekend I'll fix it). With this, if one gives it a SMILES with a star/dummy atom/R-atom, e.g. `*C(=O)CCC` for butanoyl it will make a covalent params file. It also allows the atom names to be assigned or copied from a partial match, so one can be consistent or do a ligand switcheroo. I have another module, Fragmenstein, which can be used for a ligand switcheroo (example), but if it is a one off, it's easier to remove an atom or two in PyMol.
There are three types of connections in a topology file in Rosetta. UPPER, LOWER and CONN. The first two are for polymers and the last for ligands/sidechains. So you strictly have to use CONN1 here.
For a PDB file with a covalent ligand the important thing is that there is a LINK record. PyMol strips these, but are easy to write manually.
Rosetta after subversion 10 IIRC has been good with crosslinks to aspartate —for earlier versions you need to have a special ASX residue.
One thing to mind is the fact that the x-link bond topology may be off, FastRelax with cartesian settings deals somewhat with it, but having assigned constraints is better, especially if your bond is planar as a CONNECT entry is a single bond with no resonance —not sure yours is.
The bridging oxygen, I believe needs to be part of the ligand (`*OC(=O)CCCCCC`). Note that the OG1 oxygen will become OG.
Do note the figure is not 100% correct as it depects technically a radical reaction as opposed to a bog-standard hydrolysis:
If it were a radical reaction you would need HOOH or O=O and not standard water.
So you actually have two steps, one is the stable crosslinked intermediate and the other is the hydrolysis transition step after the addition of the water nucleophile and before the elimination of one of the carboxys —this is chiral, but the hemiacetal can be modelled.
Hello,
Thank you for your response.
I am not completely sure, but I believe the second step is hydrolysis and the water molecule should be oriented correctly (by hydrogen bonding with His) to attack the anhydride intermediate from nucleophilic attack from D281 in the first step based on the mechanism of similar enzymes.
I have some questions:
1. you mentioned that "So you actually have two steps, one is the stable crosslinked intermediate and the other is the hydrolysis transition step after the addition of the water nucleophile and before the elimination of one of the carboxys —this is chiral, but the hemiacetal can be modelled."
I hypothesize it is just sufficient to model the crosslinked intermediate for studying specificity, and modeling the hydrolysis transition might not be necessary even though I am not sure. Do you have any suggestion about this?
Also, I think the crosslinked intermediate (anhydride) is chiral, but the hydrolysis transition step after the addition of water nucleophicl is achiral. Is it right?
2. I ran rdkit_to_params for `*OC(=O)CCCCCC` and obtained the parameter file attached to this forum. How I should modify this parameter file to ensure the O1 of the ligand (OG1 of D281 of enzyme) is connected to CG of D281 of the enzyme. I see there is CONN1 in the parameter file. Do I need to change that? Moreover, I am confused how the OG1 of the enzyme can be used as an atom for the ligand.
3. My plan is to get the binding score (interface score) between the ligand and different mutants of the enzyme. So I need to run ligand docking or rosetta enzyme design application. For that, is it necessary to define the link record in PDB, or defining the covalent bond as a constraint with AddorRemoveMatchCsts would be enough? any suggestion for this work is really appreciated.
Hello,
I followed your instruction: I first removed the OD2 line of ASP 281 and considered OD2 as the atom of the ligand. Then, I added the link line to the input PDB file. Finally, I performed docking (just minimizing and scoring with the constraint below) with the parameter I generated from your website, but I got a very big value for interface score and the output pdb does not show any covalent bond between the enzyme and the ligand. Please see the attached files.
If I remove the constraint mover, the interface score sounds reasonable. But I need to have the covalent bond between the ligand and the enzyme.
There is another problem: The ASP 281 in the protein has both OD1 and OD2, even though I removed OD2 in the input PDB. It seems that I need to use patch ASP. Could you guid me with making ASP patch which is connected to the ligand.
AtomPair CG 281A OD2 1X HARMONIC 1.36 0.2
Angle C3 1X OD2 1X CG 281A HARMONIC 125.9 0.35
Angle OD2 1X CG 281A OD1 281A HARMONIC 128.7 0.35
Dihedral OD1 281A CG 281A OD2 1X C3 1X CIRCULARHARMONIC -3.90 0.35
I appreciate your help and any suggestion.
I am attaching the input file, log file , consrraints , parameter and the output.
Phew! The figure is misleading then. There are cases where molecular oxygen breaks up a molecule by a radical SAM enzyme, such as a hydrocarbon chain by BioI in B. subtilis and it is very nasty/ugly stuff. Your step is a classic addition/elimination affair.
1. If you are altering the specificity without changing the mechanism, e.g. different length substrate, that is totally fine —and the data will make more sense. If you are changing the reaction from hydrolysis to transacetylation or improving k_cat as opposed to k_on/k_off, then you may need to consider it.
Chirality. An anhydride is near planar and has no hydrogens.
2. The caproyl is a Ligand and not a polymer, so CONN1 is correct. I honestly have not delt with a crosslinked aspartate where the linking atom is an oxygen, it generally is a nitrogen and there there is no OG2. The crosslinking patch of ASP in the database might support OG1, but it would strike me as odd.
So removing the line in the PDB manually is fine.
The LINK record I mentioned is a PDB format feature you add to the top of your PDB file, which automatically gets parsed by the PDB reading mover.
This adds a connection type bond with is different that a constraint as it does not result in a horrendous LJ term.
The constraint file would be something like
Hello,
Please see my post #4. I mistakenlly replied to the last but one post.
Thanks
PDB files are column-based not space-separated. The correct column-format of your LINK line is
You get loads of warning because some atom names for hydrogens differ. However, this also happens with a corrected LINK:
Along with loads of heavy atom deletions. The heavy atoms in the topology file **must** match the PDB file.
On further inspection I see that the params I linked above was for capronic acid as an example, while the ligand there is lauric acid (`*OC(=O)CCCCCCCCCCC`).
Also you have a TER line before your ligand.
In Pyrosetta at least the two files attached work fine.
Hello Matteoferla,
Thank you for the response.
I corrected the mismatch of names between pdb file and the parameter file and also added TER and link line as you mentioned. But I am still having trouble mentioned below: Note that I am working with Rosetta, not PyRosetta.
I just ran scoring mover with constraint in the ligand docking.
1. In the output, the ligand is not attached to the protein and also ASP281 has two own oxygens and ligand also has own oxygens (Please see the attached file).
2. The interface score is a very negative number. If I remove the constraint, the interface score becomes reasonable, but the first problem mentioned above exists. The correct modeling needs the constraint.
My constraint file contains the line below:
AtomPair CG 281A OD2 1X HARMONIC 1.36 0.2
my options file contains the flag below:
-packing
-ex1
-ex2
-no_optH false
-flip_HNQ true
-ignore_ligand_chi true
-overwrite
-mistakes
-restore_pre_talaris_2013_behavior true
Did I miss something or did I do something different so that my output is different from yours?
I am attaching my output file, docking xml and score file.
I really appreciate your help
I use PyRosetta and I am used to it. It runs the same C+ functions as Rosetta Scripts under the hood: an electric mini cooper and an electric BMW i3 run the same engine, but look different on the outside and are driven by folk with different likes. So what works for one, should work for the other. I frequently use the tests in the source code to use PyRosetta code.
I meant the opposite about the TER, but I said a porkie pie (lie). It makes no difference here. I think it is a problem with Remodel or something.
Just now I downloaded your files. I added the aforementioned LINK line before the coordinates. I deleted 182:A.OD2. I renamed OD1 to OD and a space for correct tabulation (important). The isopeptide is there. Could you try dumping the PDB right after loading? The link entry should be there —PyMOL respects it, but NGL.js based web tools requires CONECT for LINK.
I have not used InterfaceBuilder, so I am not sure what you mean about negative being bad: I am assuming its not a potential (down the energy well = happy)? But constraints do generally penalise and will give slightly worse scores —but preferably negligible.
Hello,
I am attaching the score file and log file and the input PDB file. The interface score is -6131968.450. I used the same PDB file in post #7 (you gave me) with only renaming the ligand atoms to be consistent with the parameter file.
I used the command below for runnin Rosetta:
rosetta_scripts.static.linuxgccrelease @options.txt -in:file:s ../../input_files/BTE_C12_Rosetta_forums_rename.pdb -in:file:extra_res_fa ../../input_files/LAU.params -qsar:grid_dir ./C12_score_ter_July9_minimize_w_constraint_grid/ -out:nstruct 1 > out.log
If something is wrong in my simulation, please let me know.
Thanks
Okay, on my system (running release r247 2020.08+release), I tried the attached files in a blank script
It appear to be fine on my system. Therefore one of your options is not playing nice with the crosslink. Could you make a blank XML and test your protein and slowly add your options?
Alternatively there is the crosslinked Aspartate in the database. To use this you need to change ASP to ASX in the LINK and in the ATOM records of your PDB —and revert back OD to OD1. To work it needs the LINK record just like the regular ASP —so there should be no need for this extra step... but it's not working, so hey.
However, there is another problem. Six million kcal/mol —uranium fission is a billion kcal/mol, so not quite but nearly there. Your interface energy should not be lower than that of your complex. The interface energy is generally calculated by pulling the chains apart. This does not break the crosslink. Therefore your interface is comically favourable because it's pulling your crosslink like an elestic band —it's irreversibly bond from the forcefield's point of view. This is however at odds with the fact your script is killing your crosslink...
Hello Matteoferla,
I would like to thank you for your help. ASX worked for me. I also solved the problem with that million kcal/mol interface. Previously, I considered the constraint as a mover before minimization. But I realized if I considered the constraint as an option, the interface score would be reasonable.
I have another question: The output of the above simulation has a distance of 2.2 between CG of ASX and ligand Oxygen(which was previously OD2 of ASP), while in the input it was 1.36 and I made the constraint with 0.2 standard deviations as below. I increased the constraint weight to 100, but it did not help. I was wondering whether this issue can be solved. Is there any reason that Rosetta cannot decrease this distance?
AtomPair CG 281A OD2 1X HARMONIC 1.36 0.2
I am attaching the output PDB.
Glad you are getting there!
Many reasons for issue. Please look at the scored per-residue values at the bottom of the PDB files before and after minimisation.
label fa_atr fa_rep fa_sol fa_dun
SPE_277 -9.55207 0.87345 4.31866 0
ASP:SidechainConjugation_192 -3.94005 0.43509 4.08479 6.71051
SPE_277 -5.59201 0.31076 1.92523 0
ASX_192 -3.26199 0.23884 1.49105 1.27095
The solvatation term is large —many hydrophobics in the solvent beforehand?— and the sidechain internal energy (a statistical term) deviates from Dunbrack's values. However, 100 *((2.2 - 1.32)/0.2)**2 is gonna be like 2,500 kcal/mol. Looking at the file you gave I see:
angle_constraint 0
atom_pair_constraint 0
chainbreak 0
classic_grid_X -37
coordinate_constraint 6.9246
dihedral_constraint 0
Your atom_pair_constraint did not stick —could you double check the reweight entry in your scorefunction is spelt right? I for one cannot spell comstraight.
【Note to future folk sent hither by The Google overlord: ASP with the side conjugation patch is preferable to ASX, so please try that first】