
Hello,
I usually use my NCAAs by mutation in the sequences.
Now, I am looking for a way or application in Rosetta to add, not mutation, NCAAs to the N- terminal and C- terminal of the sequences.
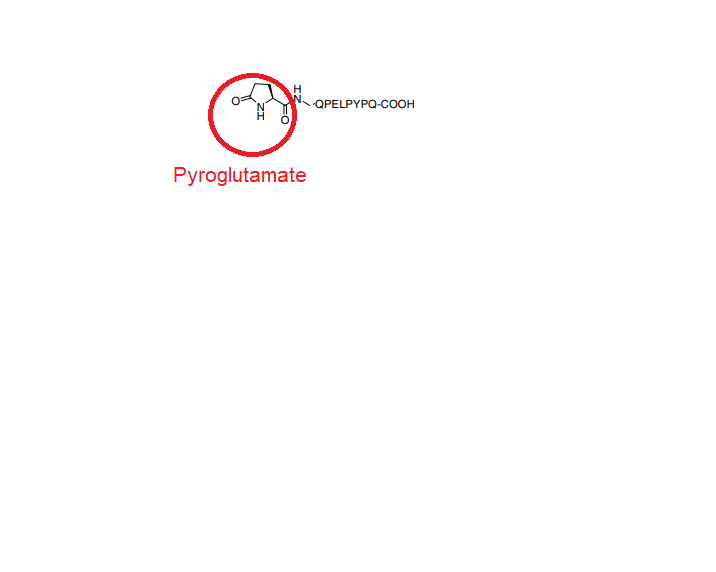
The RCSB pdb starts with leu at N- terminal in attached picture that we need to add pyroglutamate before that
I wonder if anyone could let me know how I can add pyroglutamate using params file and rotlib file to the sequence.
| Attachment | Size |
|---|---|
| 9.56 KB |
{kind=link}
Category:
Post Situation:
A terminal residue in a params file is straightforward and is just a regular polymer residue with three differences.
However, just like proline, you don't need to make it a N-terminal cap and it can be a regular residue with a regular hydrogen as a cap.
rotlib file --> surely you want solely the cis or the trans rotamer, so you have a single rotamer, so all is good.
Something like this (assuming I got L and trans right)
Thank you for your explanation and example file Could you please let me know how I can add the ncaa to n-terminal of pdb file
I mean, What script or application should be used
I normally use Pyrosetta for everything and do not know how one could do it with Rosetta Scripts.
One crude option is to add a proline in PyMOL by selecting the pre-cap residue's N atom (editor mode), then menu -> builder -> add proline, and the making that wretched amino acid `GLP` or whatever you want to call it (has to match the topology file) by typing `alter chain X and resi xx, "resn='GLP'"` and `sort`, where xx and X are residue index and chain. Rosetta when reading this file will have a fit and the wrong atom names —but the first three atoms match so will fix it as it does for missing sidechains (but not for C-alpha traces). As I said, this is really nasty. An alternative is loading a file with GLP and an amino acid and aligning it to say a temporary residue build at the N-terminal or aligned via a second residue on GLP (see below).
The topology file is fine, except for me I had to delete `LOWER_TERMINUS_VARIANT` from the params file, so `TERMINUS` is all that is required, property-wise, sorry for the incorrect info!
But doing the following gives me the attached figure. I am not suggesting switching to PyRosetta as I think regular Rosetta may be able to do what you hope.
Which makes me realise that the cys/trans business does not apply to N-terminal residues —ops again.
The PDB block of the first two residues looks like:
This can be used for alignments. Actually, even with PyRosetta you'd make a dipeptide with GLP and align that to the pose before removing the second residue, removing the terminal cap on the proper protein pose and appending the latter to the former —with some code to copy along the PDBInfo.
I am sorry. I do not have any help, advice or solutions to offer as I am very new to Rosetta.
Could you share with me any material or literature that goes into detail and could guide me in mutating canonical amino acids in peptides to NCAA (such as D amino acids, AIB etc.) ?
Thank you.
Email ID - subhrodeeps@iisc.ac.in