
Hello, friends,
I was designing the "interface" (possible contacts) of a fusion protein using FastDesign. Since I was running the appliction on a laptop, so far only 200 designed models have been genrated.
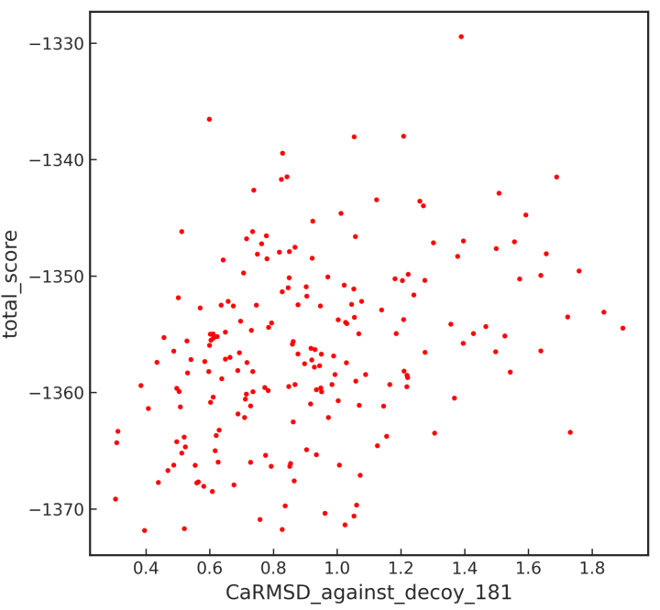
I then went on to analyze the result anyway and tried to plot Score VS caRMSD (as shown in the attached file). Here are my questions:
(1) When plotting Score VS caRMSD, i chose the decoy with the lowest total score as the reference to calculate RMSD against. Is this reasonable? if not, what structure should i choose as the referece? The fusion protein model before design is generated by FastRelax and i don't think it's a "native" structure.
(2) Dose the current Score VS caRMSD plot look good or show some degree of convergence? Or i have to generate more decoys so that i can tell the degree of convergence? And how many decoys should be generated? I heared from some authors that they sometimes only generate one designed sequence and just go on without extensive design.
(3) This may sound naive but i am wondering when i choose the model with lowest score as the referecen and plot the Score VS RMSD, which structure should i choose as the best one? Isn't the reference decoy the best one?
Thank you very much
Yours sincerely,
lanmei
| Attachment | Size |
|---|---|
| 68.08 KB |
{kind=link}
Hi,
1) Score vs RMSD is most useful for denovo folding of one sequence. I.e trying to get to your design from an extended backbone. Honestly today I would just use alphafold and check how well the structure is predicted.
2) No convergence, but just take the lowes score, if you do not have target constraitns.
3) All things being equal lower is better. But also pay attention to pI, charge, packing quality etc...
Best,
Ajasja
Hi, Ajasja,
Thank you for your reply.
I agree that AlphaFold is a good choice to check my fusion proteins. And I am going to select the model of lowest socre and check other parameters as you said.
Yours sincerely,
lanmei