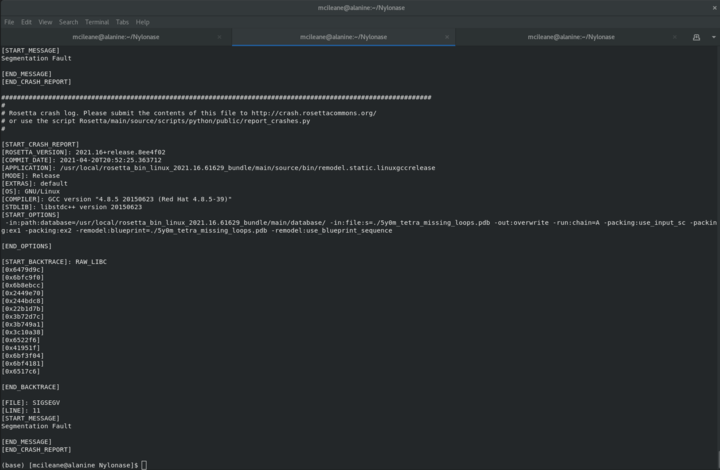
Segmentation Fault
Submitted by ileanexis on Mon, 2024-03-11 08:53
Category:
Loop Modeling
Hello! I'm trying to run add some missing loop to my protein by appear segmentation fault as error.
Anyone have an idea to solve this issue?
_________________________________________________________________________________________________________________________________________________________________________________
Post Situation:
Forums:
- Read more about Segmentation Fault
- Log in or register to post comments