
Hi there,
I am trying to relax my structure which contains a bound ligand.
First, I created a LG.params file using:
python /path/to/molfile_to_params.py /path/to/1XKK_ligand.mol2
This file (1xKK_ligand.mol2) only contains the coordinates for the ligand from the PDB 1XKK. molfile_to_params.py outputs the LG.params file as well as a PDB, which looks correct when visualy inspected. This is all expected.
Then I am running the relax application with the following command:
relax.macosclangrelease \
-database $ROSETTA_DATABASE \
-s /path/to/1XKK.clean.pdb \
-in:file:fullatom \
-native /path/to/1XKK.clean.pdb \
-out:file:silent 1XKK.relaxed.a.out \
-nstruct 1 \
-out:file:scorefile 1XKK.relaxed.a.score.csv \
-extra_res_fa /path/to/FMM.params \
-relax:constrain_relax_to_start_coords \
-relax:coord_constrain_sidechains \
-relax:ramp_constraints false \
-ex1 \
-ex2 \
-use_input_sc \
-correct \
-no_his_his_pairE \
-no_optH false \
-flip_HNQ
This command executes ok, but when I look at the final PDB after extracting it from the silent output file, I see that the coordinates for the ligand are completely messed up. The side chains for the protein itself look ok (not shown in the images below).
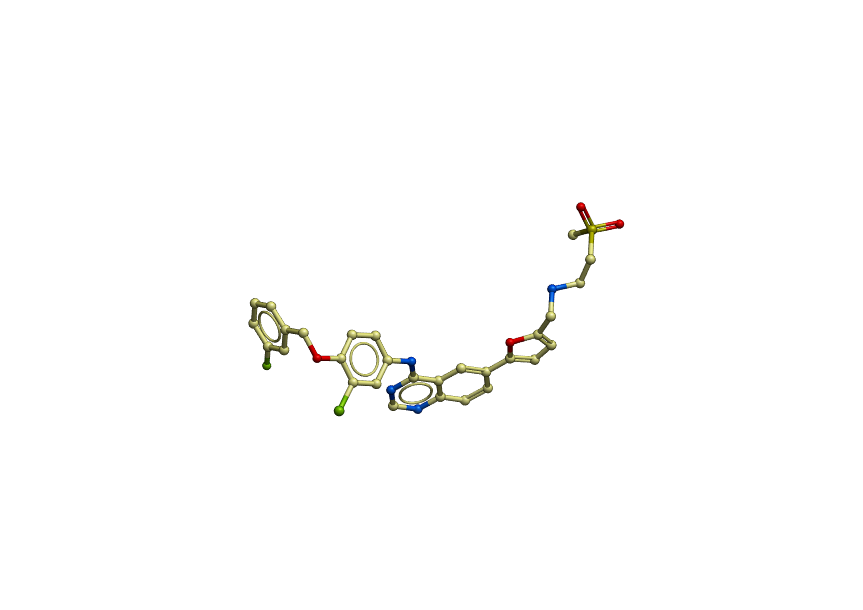
Here are what the CORRECT (before relax) ligand looks like:
https://dl.dropboxusercontent.com/u/4402291/pdb/ligand_correct.png
{kind=link}
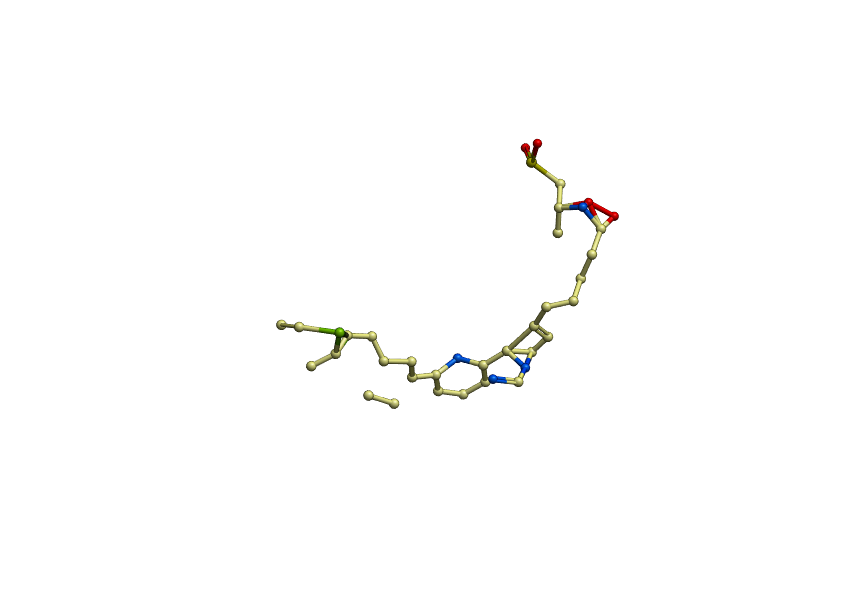
And here is what the INCORRECT (after relax) ligand looke like:
https://dl.dropboxusercontent.com/u/4402291/pdb/ligand_incorrect.png
{kind=link}
Does anyone have any idea why this is happening???
Many thanks!
Brett
<!--break-->
| Attachment | Size |
|---|---|
| 30.58 KB | |
| 26.26 KB |
{kind=link}
{kind=link}
The atom names of the ligand are very important - that's how Rosetta knows which atoms in the PDB match up with which atoms in the params file. What you're showing looks very much like what might happen if the atom names get reorganized.
By default, molfile_to_params.py often will rename atoms, so if you're using a PDB file with a ligand created prior to your molfile_to_params step, the atom names might not match. The "--keep-names" option to molfile_to_params sometimes helps, but it may have to rename atoms anyway, if there's duplicates, or if your input mol/mol2 file doesn't have atom name information. In those cases, you'll want to use the PDB file that molfile_to_params produces - it should have the same coordinates, but with the param file atom names.
I'll also note that on recent weekly releases there is an experimental option on Rosetta itself, invoked by "-remap_pdb_atom_names_for <three letter code>", which would cause Rosetta to ignore the atom names on the input PDB for the given residue, and instead use structural heuristics to determine how atoms match up. This won't always work well for all ligands, however, so be forewarned.