
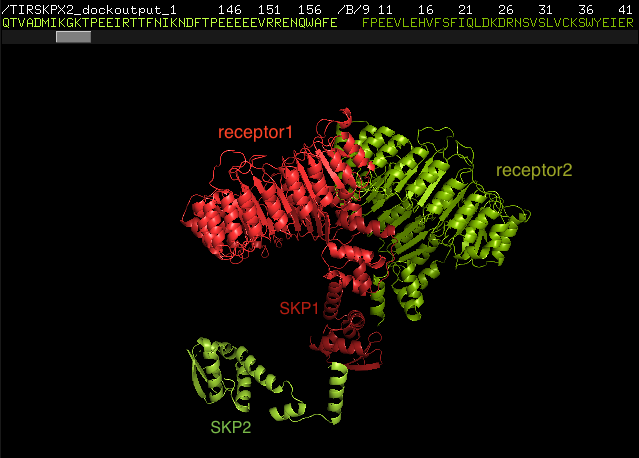
We want to dock 2 proteins (red and green) to see how they dimerize. Each crystal structure has 2 parts that are not connected to one another (The receptor at the top and the SKP part below do not have any linkers to make the 2 parts become one chain) They just interact with some electrostatic interactions. This makes the receptor and SKP apart from each other for all docking outputs. From the literature review, we could not reject the SKP part on docking because it has some effect on receptor conformation. How can we constrain this docking problem, since we know some of the interacting residues?
PS. I have tried to get the receptor and SKP on the same chain but the structure was broken. I have also tried to specify the relative chain partners as "AC_BD" (set both SKP parts to be rigid, and receptors to be flexible), but it did not work.
| Attachment | Size |
|---|---|
| 109.2 KB |
{kind=link}
Why specifically did "AC_BD" fail? Is everything on the same segment (`segi` in PyMOL)?
Constraints for Rosetta are documented here: https://www.rosettacommons.org/docs/latest/rosetta_basics/file_types/constraint-file
They are technically "restraints" not constraints as they are penalties as opposed to keeping things absolutely fixed. If you want something fixed in an AMBER style "constraint", you need to play with movemaps or packers.
For pyrosetta there are three ways to specify constraints:
From file
From a set of constraints.
From a generator and a residue selector
Add weights
A constraint is added to a pose, but the weight of a constraint is in a scorefunction. This is true for the binaries and scripts, where the weight has to be specified.
Centroid
One thing to note is that the scorefunction for the centroid mode also needs the weights, but the centroid residues have a beta carbon only as a sidechain. So changes may be required. Docking protocols go through a centroid mode.