Stub Libraries for DockWithHotspotMover
Submitted by ayush_patel on Sat, 2024-03-09 22:43
Category:
Docking
Hello!
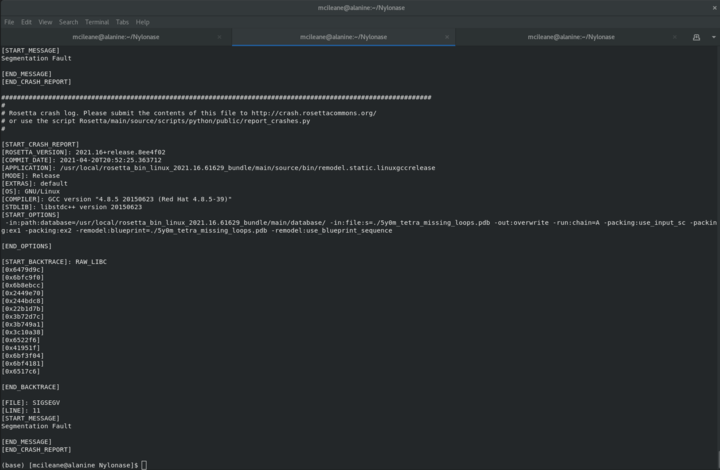
I was a little bit confused about what exactly the stub libraries are and how we are supposed to generate them. From what I understand, they're simply just disembodied residues that we consider "important" to the interface in the docked complex. So, are the stub files just generated by taking a protein structure and deleting everything except for the hotspot residue, then saving that singular residue as a PDB file? Also, is it possible to assign hotspot residues to both chains that we are docking together?
Thank you so much.
Post Situation:
Forums:
- Read more about Stub Libraries for DockWithHotspotMover
- Log in or register to post comments