Biohub releases open source models that utilize evolution to design disease target binders
Key Takeaways
- Biohub has released open source ESM protein language models that use evolutionary data to streamline drug discovery and map functional protein spaces.
- ESMFold2 successfully generated lab-validated, high-affinity protein binders against five disease targets in cancer and immunology.
- The models were used to generate the ESM Atlas, which maps 6.8 billion sequences and 1.1 billion predicted structures to protein functions.
Biohub, a non-profit research organization started by Priscilla Chan and Mark Zuckerberg, has released updates to open source ESM protein language models after recruiting the team from EvolutionaryScale. Alex Rives, Ph.D., one of the keynote speakers for Summer RosettaCon 2026, is head of science at Biohub and was the former chief scientist at EvolutionaryScale. Read a summary about the release below and check out the preprint for more details.
Translating evolutionary data into structures at scale
Researchers at Biohub are working to streamline the process of identifying viable drug candidates through AI-based predictions of protein design. Through their research, Biohub has unveiled the latest updates to the ESM protein language model family. The system includes ESMC (Evolutionary Scale Modeling Cambrian), a language model trained on approximately 2.8 billion sequences from a wide variety of life, including organisms from extreme environments and over 20,000 human proteins. To convert this evolutionary information into physical structures, the system pairs ESMC with ESMFold2, a prediction model and design engine. Instead of requiring multiple sequence alignments to build representations, ESMFold2 captures information learned during its pretraining. It also features a looped transformer architecture that helps scale compute during inference and prevents overfitting.
Designing binders for disease targets
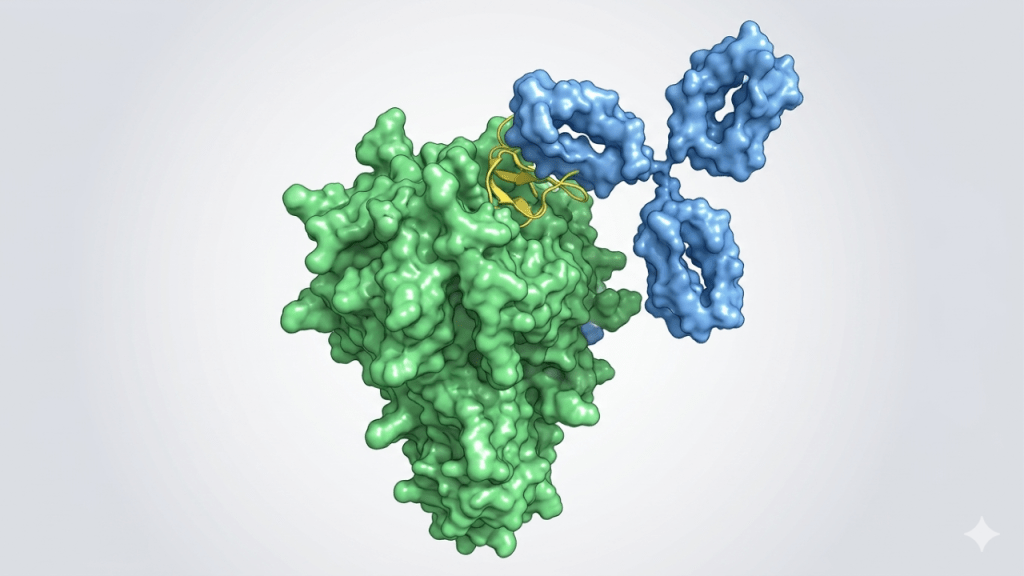
The updated ESM models have expanded capabilities in designing binders and mapping protein functions. Biohub used ESMFold2 to generate high-affinity protein binders against five specific disease targets in cancer and immunology. These targets included receptor tyrosine kinases (EGFR and PDGFRβ), immune checkpoints (PD-L1 and CTLA-4), and a regulator of immune cell signaling (CD45). In performance benchmarks, ESMFold2 compared favorably against other models, including Chai-1, Boltz-1, and AlphaFold 3.
Laboratory testing validated the performance of these automated designs. The generated mini-binders achieved hit rates between 36% and 88%, while antibody-derived formats achieved hit rates between 15% and 29%. The binders demonstrated nanomolar binding affinity, high specificity, and favorable stability profiles. Furthermore, tests showed that the binders designed for PD-L1 restored T-cell signaling in the laboratory by successfully blocking the same pathway targeted by approved checkpoint therapies.
Mapping the protein universe
Beyond individual binders, Biohub applied the models to generate the ESM Atlas, which maps 6.8 billion sequences and 1.1 billion predicted structures to protein functions. By analyzing the model’s internal representations with sparse autoencoders, researchers discovered that the system independently learned hierarchical concepts, ranging from basic amino acid chemistry to functional concepts across unrelated proteins. Notably, the atlas clustered together highly divergent proteins with low sequence similarity, such as RNA-guided DNA endonucleases, eukaryotic Fanzor proteins, and prokaryotic TnpB. These specific insights could eventually support the creation of new gene-editing tools.