Science Updates
Debating the balance between model openness and biosecurity guardrails

Key Takeaways The scientific community is currently debating how to balance open research with biosecurity protections when it comes to biological AI tools. Biological AI tools are primarily designed for drug discovery, treating illnesses, and other broader societal benefits. Advancements in AI, such as chatbots, could provide opportunities for even inexperienced bad actors to misuse…
Read MoreBiohub releases open source models that utilize evolution to design disease target binders
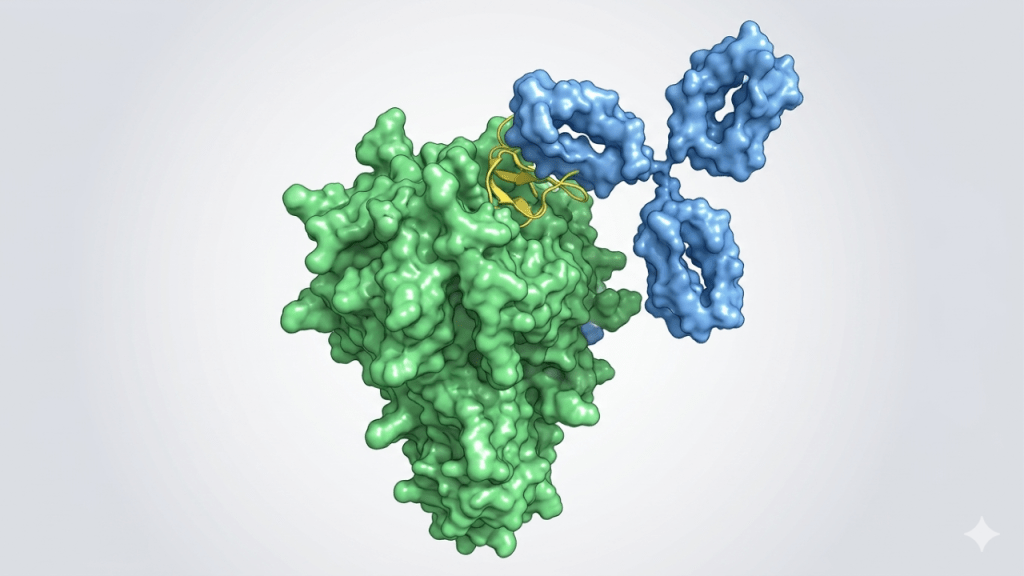
Key Takeaways Biohub has released open source ESM protein language models that use evolutionary data to streamline drug discovery and map functional protein spaces. ESMFold2 successfully generated lab-validated, high-affinity protein binders against five disease targets in cancer and immunology. The models were used to generate the ESM Atlas, which maps 6.8 billion sequences and 1.1…
Read MoreOpenBind releases initial structure and affinity dataset for structure-based machine learning
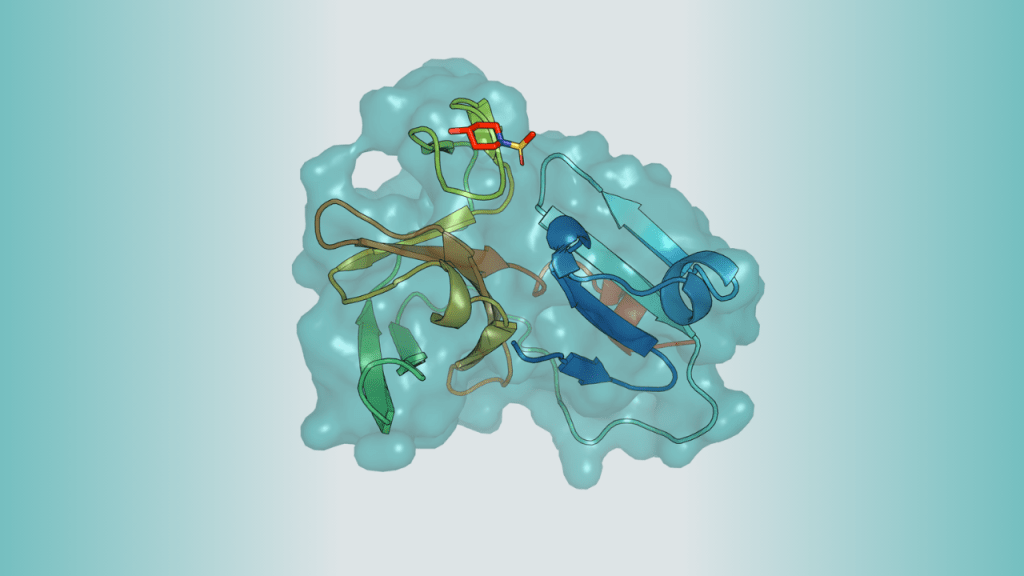
Key Takeaways OpenBind is generating dense, high-quality protein-ligand datasets that link structural data with binding measurements at a large scale. This release provides a realistic, structurally novel testbed linking high-resolution crystallographic data directly with biophysical binding affinity. The initial release targets the Enterovirus A71 (EV-A71) 2A protease (modeled via a closely related Coxsackievirus A16 surrogate…
Read MoreSpeed Up PyRosetta Development with Autocompletion and Type Checking

By Harrison Truscott PyRosetta, the Python interface to the Rosetta binary, now comes packaged with type stub files (.pyi). These files describe the module’s classes, methods, and variables, as well as function signatures, types of function parameters and return values, and descriptive docstrings. Type stub files can be used by the Integrated Development Environment (IDE)…
Read MoreA Zero-Shot Approach to de novo Metalloenzyme Design
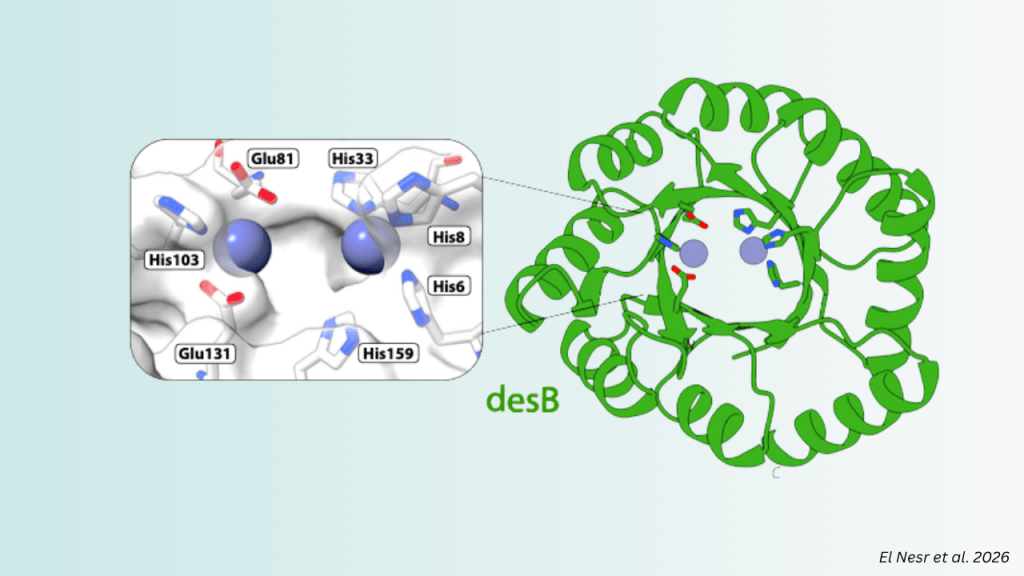
Key Takeaways: dEVA (design by EVolutionary Algorithm) is a framework that achieves the zero-shot design of a highly efficient metalloenzyme without any reliance on natural templates. Design objectives were tailored to the specific chemistry of metalloenzymes, in particular catalytic zinc sites, and from only three experimentally-tested designs found desB, the most efficient de novo designed…
Read MoreDeep Learning’s Impact on de novo Protein Design
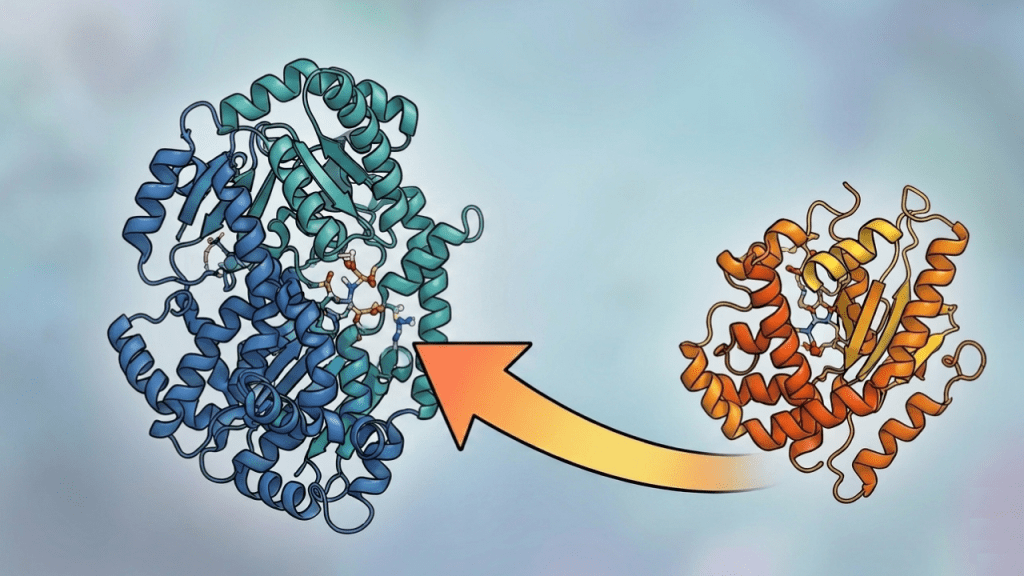
Key Takeaways: With advancements in AI, the field of protein engineering has shifted from template-based approaches to de novo designs with approaches including seed-based design, deep generative models, and binder hallucination. Design towards specific protein functions is the current frontier factoring in parameters including neosurfaces, conditional binding with biological stimuli, and dynamic modeling, linking deep…
Read MoreNext Generation Generative Model Unlocks de novo Designs at Scale

Key Takeaways: NVIDIA’s new Proteina-Complexa model combines generative AI with inference-time search algorithms to operate 30-60x faster than RFdiffusion when designing custom proteins that target specific diseases. In a test to find binders for 127 different targets from over 1 million protein designs, the model successfully found binders for 68% of the targets. The model…
Read MoreRosetta Commons Run (rc-run) Now Available

We are pleased to announce a new Rosetta Commons workflow optimization tool: rc-run. rc-run (invoked as rc) is a command-line utility tool for running containerized and native biomolecular software. It simplifies common tasks, such as mounting local directories, building HPC containers, running containerized applications, and logging executed commands, to make complex workflows easier to reliably…
Read MoreCyclicMPNN adapts sequence design to cyclic backbones

Why this work matters Cyclic peptides are attractive scaffolds because their closed backbone can promote structural rigidity and proteolytic stability. Yet assigning sequences that reliably fold into a given cyclic backbone remains a key challenge. In this study, the authors introduce CyclicMPNN, a fine-tuned version of ProteinMPNN designed specifically to improve sequence design for short…
Read MoreFoundry Docker Image Now Available

Rosetta Commons now offers an official Foundry Docker image designed to make it dramatically easier to run cutting-edge biomolecular machine learning models without complex setup. Foundry is the central repository for a suite of ML models used in protein science—including RF3 (a next-generation biomolecular structure prediction network), ProteinMPNN inverse-folding models, and the newest RFdiffusion3 (RFD3)…
Read More